

Version 1.0
The samples that PASS the pre-aggregation QC pipeline (per sample QC) are aggregated into one multi-sample VCF file. Samples are aggregated using Dragen Iterative gVCF genotyper. To accelerate the aggregation and the downstream analyses (parallel processing), the genome is divided into 100 shards.
The pipeline produces one multi-sample VCF file per shard, as well as a global multi-sample VCF file that concatenates all shards.
Alongside the multi-sample VCF generation, we also perform the following analyses:
Functional annotation
Each msVCF (global or per shard) is annotated using Ensembl Variant Effect Predictor (VEP) tool, version 112. In addition to VEP default annotations, we added the below databases:
- ClinVar v20240805
- HGMD Pro v2024.2
- GnomAD genomes v4.0
- GnomAD exomes v4.0
High quality dataset of SNPs (HQ) - (cohort-level QC)
We initially converted the multi-sample VCF (msVCF) into Bed/Bim/Fam format using the Plink 1.9 tool. These files were then utilized to generate a high-quality set of SNPs, also using Plink 1.9. The SNPs were selected based on the following criteria:
- Include autosomal, bi-allelic SNPs only
- Common variants only (MAF > 5%)
- Remove variants with missingness rate > 10%
- Remove samples with missingness rate > 5%
- LD prune with an r2 of 0.1, 500kb window
- Hardy Weinberg Equilibrium pHWE < 1e-5
Principal Components and genetically inferred relatedness
Using the HQ SNPs, we run PCA using the GCTA tool (v1.94). The first 20 PCs are provided. We also generated a pairwise kinship matrix using KING tool (v2.2.7), in addition to a list of unrelated samples (up to 2nd-degree).
Hail matrix
The global msVCF, is also converted into Hail (v0.2) MatrixTable format. This matrix can be imported to perform highly parallelized analysis.
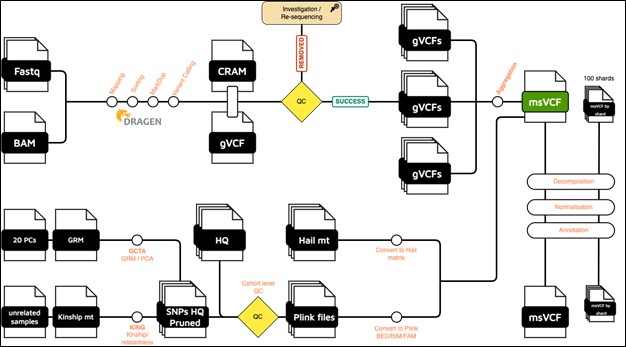
Figure 1: Global overview of the QPHI analysis pipeline